
28. FOETAL UROLOGY
28.1. Normal renal development
28.1.1. Introduction
Congenital urogenital anomalies account for approximately 20-30% of all abnormalities detected on prenatal ultrasonography, with urinary tract malformations being the most frequent within this group [1584-1588]. These anomalies range from mild findings, such as low-grade hydronephrosis, to conditions incompatible with life, such as bilateral renal agenesis. Over recent decades, prenatal diagnosis has advanced considerably due to the widespread use of second- and third-trimester ultrasonography and, increasingly, foetal magnetic resonance imaging as a complementary tool. Early detection enables delivery planning in tertiary centres with multidisciplinary teams. It also allows tailored postnatal management aimed at reducing neonatal morbidity and optimising long-term renal prognosis. Accurate interpretation of congenital anomalies requires knowledge of normal urinary tract embryology and the principal aspects of imaging assessment. Prenatal findings provide essential information for parental counselling and perinatal planning. Families should be informed not only about the immediate anatomical findings but also about the possible impact on renal and/or bladder function, associations with genetic or syndromic conditions, and implications for neonatal management [1589,1590]. This highlights the role of prenatal imaging as both a diagnostic and a prognostic tool, while underscoring its limitations. At the same time, it is important to communicate that prenatal diagnoses are not necessarily definitive and may require postnatal confirmation to establish the exact nature and severity of the condition.
28.1.2. Renal and bladder development
28.1.2.a. Normal development
The kidneys develop from the intermediate mesoderm through three successive stages, with the metanephros forming the permanent kidney. Migration from the pelvis to the lumbar region occurs between weeks 6 and 9, and abnormal interaction or ascent may result in ectopia, fusion anomalies such as horseshoe kidney, or renal agenesis [1589,1591,1592]. Complete failure of ureteric bud induction may result in unilateral or bilateral renal agenesis [1591]. Persistence of embryonic vessels may lead to accessory renal arteries, which in some cases can contribute to ureteropelvic junction obstruction [1592].
Corticomedullary differentiation becomes progressively more visible with advancing gestation and should be routinely assessed during prenatal imaging [1589].
The urinary bladder originates from the anterior cloaca and can usually be identified from 9 to 10 weeks as a midline anechoic structure that fills and empties regularly. The visualisation of the urinary bladder confirms the presence of at least one functioning kidney and offers an early indirect marker of urinary tract patency [1589].
Understanding normal renal and bladder development is essential for interpreting their appearance on prenatal imaging.
28.1.2.b. Prenatal ultrasound
Prenatal ultrasound is an essential technique for monitoring foetal development and detecting congenital anomalies. Although the frequency and timing of these examinations vary between countries, there is broad consensus that all pregnant women should undergo at least one ultrasound in the first trimester and another in the second trimester, while the use of a third-trimester scan depends on national recommendations.
While ultrasound remains the cornerstone for detection of urinary tract malformations, its accuracy can be limited by maternal factors, oligohydramnios or when complex malformations are suspected. In such situations, foetal MRI can provide complementary information to better define anatomy and guide counselling [1593].
• First trimester (11-14 weeks)
During this period, the main objectives of ultrasound are to confirm pregnancy viability, establish accurate gestational age and initiate screening for aneuploidy and early preeclampsia [1594-1596]. In addition, first-trimester ultrasound can identify some major structural anomalies. Early recognition of such findings enables earlier genetic testing and provides more time for parental counselling and decision-making. When an anomaly is detected or suspected at this stage, the patient should be referred to a specialised centre without waiting for the midtrimester scan [1589,1590].
The urinary bladder should always be visible from 12 weeks onwards with a longitudinal diameter < 7mm [1228,1594]. Persistent no visualisation at this stage may suggest bilateral renal agenesis, cloacal exstrophy or bladder exstrophy, and requires repeat scanning for confirmation. The kidneys are usually detectable from 12 to 13 weeks in a paravertebral position, with reniform morphology and a central hypoechoic pelvis. The echogenicity of kidneys is compared to that of the liver and spleen. Ureters are not normally visible [1589].
The most characteristic urinary malformation detectable in this period is megacystis, which is defined as a bladder diameter ≥ 7mm between 11 and 14 weeks. If the bladder diameter measures between 7 and 15mm, this is considered mild megacystis. Such cases carry an increased risk of aneuploidy, but a spontaneous resolution is likely. For this reason, a follow-up ultrasound within one week is recommended to confirm progression or resolution [1590,1596]. If the diameter exceeds 15mm, the probability of significant lower urinary tract obstruction (CLUTO) is considered high. In these cases, the bladder is markedly enlarged and the ‘keyhole sign’ (distension of the proximal urethra) may be seen, with secondary effects on the kidneys (enlargement and increased echogenicity) and dilatation of the ureters [1589,1590].
Although amniotic fluid depends primarily on maternal origin in the first trimester and does not yet reflect foetal renal function, severe forms of LUTO can later evolve into oligohydramnios and pulmonary hypoplasia, which carry major prognostic implications [1589].
• Second trimester (18-22 weeks)
The second trimester represents the period of highest diagnostic yield for structural anomalies. The mid-trimester scan, performed between 18 and 22 weeks, is the key morphological assessment and allows detection of most clinically relevant urinary tract anomalies [869,1585,1590]. At this stage, amniotic fluid volume mainly reflects foetal renal function, serving as an indirect functional marker.
Normal sonographic findings
Both kidneys should be bilaterally visible in the paravertebral region, with reniform morphology and clear corticomedullary differentiation. Echogenicity should be equal to or lower than that of the liver; diffuse hyperechogenicity suggests dysplasia. The renal pelvis is physiologically < 4mm in the second trimester, whereas an anteroposterior diameter of ≥ 7mm is pathological [869,1590]. Kidney length correlates with gestational age, although longitudinal growth in growth-restricted foetuses may remain normal while transverse and anteroposterior dimensions are reduced [1585].
The urinary bladder should always be present, showing filling and emptying cycles. Longitudinal measurements average ± 14mm at 18 weeks and ± 23mm at 22 weeks [1597]. The urinary bladder should not extend above the umbilical cord insertion. Persistent nonvisualisation requires exclusion of severe anomalies such as bladder or cloacal exstrophy. The umbilical cord insertion site and the presence of two umbilical arteries should be documented - a single umbilical artery, although nonspecific, has been associated with complex anomalies [1228].
Key urological anomalies:
- Urinary tract dilatation: most frequent abnormality. Anteroposterior diameter (APD) ≥ 7mm is pathological and requires follow-up.
- Persistent megacystis: persistence beyond the first trimester almost always indicates CLUTO. The bladder is markedly distended and fails to empty, often with a ‘keyhole sign.’ Secondary findings include enlarged hyperechoic kidneys, ureteral dilatation and ultimately oligohydramnios [1589,1590,1596].
- Renal agenesis: unilateral cases present with normal bladder with filling and emptying cycles and amniotic fluid. Bilateral agenesis leads to absent bladder and kidneys, progressive anhydramnios after 20 weeks, and a universally poor prognosis.
- Multicystic dysplastic kidney (MCDK): multiple noncommunicating cysts replace normal parenchyma. Bilateral cases or those associated with contralateral agenesis have poor outcome, while unilateral cases with a normal opposite kidney usually evolve favourably.
- Diffuse renal dysplasia: diffuse hyperechogenicity with loss of corticomedullary differentiation.
- Polycystic kidney disease:
- ARPKD: massively enlarged, hyperechoic kidneys and early renal failure.
- ADPKD: enlarged, hyperechoic kidneys, usually with normal amniotic fluid, and postnatal manifestation. - Duplex collecting system and ureterocoele: duplication anomalies may occasionally be seen. A ureterocoele appears as a cystic lesion within the bladder and is highly suggestive.
- Isolated renal cysts: simple anechoic cysts within otherwise normal kidneys, generally with favourable prognosis.
- Visible ureteral dilatation: always pathological, indicating obstruction or reflux.
• Third trimester (30-32 weeks)
The third-trimester ultrasound aims to monitor previously identified findings and detect anomalies that may only become apparent at this stage [1585].
Normal findings
Kidneys are normally reniform in shape and located in the paravertebral region.
Corticomedullary differentiation becomes clearer and more consistent than in the second trimester, reflecting progressive parenchymal maturation. On ultrasound, the cortex has intermediate echogenicity compared to the liver, the medulla is more hypoechoic, and the renal pelvis appears as a central hypoechoic zone.
An anteroposterior renal pelvic diameter (APRPD) of < 7mm is considered normal, while ≥ 7mm is abnormal. An APRPD of < 15mm carries a low risk for the need for postnatal intervention, and the risk for a urological condition increases with severity of the dilatation above this cut-off [867,1598,1599].
The urinary bladder should always be visible, showing filling and emptying cycles, without wall thickening.
Amniotic fluid volume becomes a key indirect marker of foetal renal function.
Key urological findings in late gestation:
Third-trimester hydronephrosis: approximately 60% of cases are first detected during this period. Prognosis is poorer when oligohydramnios is present [1590].
- Renal cysts and dysplasia: better defined at this stage, presenting as diffuse hyperechogenicity, thinned parenchyma, and multiple noncommunicating cysts replacing the normal renal structure [1585,1590].
- Lower urinary tract obstruction: can be reassessed to determine progression. Lower urinary tract obstruction presents with a massively distended bladder with thickened wall, bilateral ureteral dilatation, blow-out of the urinary tract, and in some cases, oligohydramnios [1596,1600].
- Autosomal dominant polycystic kidney disease (ADPKD): the third trimester is the most common period of prenatal presentation. Kidneys are enlarged, with variable echogenicity and altered corticomedullary differentiation. Cysts may or may not be visualised [1585,1590].
28.1.2.c. Foetal magnetic resonance imaging
- Indications: Foetal MRI is indicated when ultrasound findings are inconclusive or when there is suspicion of a complex anomaly that cannot be fully characterised sonographically. Foetal MRI is therefore considered a complementary technique rather than a screening tool [1593,1601].
- Timing: The diagnostic performance of MRI is limited before 18-20 weeks of gestation. The optimal window is between 26 and 32 weeks, when renal parenchyma, cysts and dilatations are more easily assessed and pulmonary development can also be evaluated [1593].
- Normal appearance: On T2-weighted sequences, normal kidneys appear as reniform structures of intermediate signal intensity with a central hypointense zone corresponding to the renal pelvis. Corticomedullary differentiation becomes progressively evident as gestation advances. Renal cysts are visualised as well-defined hyperintense lesions. The urinary bladder is identified as a midline hyperintense spherical or ovoid structure with changes in size according to filling and emptying cycles. Normal ureters are generally not visible; their detection usually indicates pathological dilatation [1601,1602].
- Safety: Foetal MRI is considered safe during pregnancy when performed at the magnetic field strengths routinely used in clinical practice, with no evidence of adverse foetal effects in large observational series [1603]. The administration of gadolinium-based contrast agents is contraindicated, as gadolinium crosses the placenta and may accumulate in amniotic fluid and foetal tissues, raising concerns about potential teratogenic and toxic effects. In addition, maternal anxiety or emotional distress during the examination has been reported, underscoring the importance of adequate pre-test counselling and support [1604].
- Diagnostic contribution: Several studies have demonstrated that MRI can provide decisive information that directly impacts prenatal counselling and management:
- Differentiation of multicystic dysplastic kidney (MCDK) from severe hydronephrosis [1593].
- Characterisation of abdominal wall and pelvic anomalies, including the bladder exstrophy-epispadias complex, when ultrasound is inconclusive [1605-1607].
- Evaluation of pulmonary volume and morphology in severe lower urinary tract obstruction (CLUTO) or prune belly syndrome, where prognosis depends on both renal and pulmonary status [1608,1609].
- Distinction between unilateral MCDK and bilateral cystic kidney disease, an important determinant of postnatal prognosis [1602,1610].
28.1.2.d. Genetic counselling and multidisciplinary approach
This section addresses the key questions related to genetic counselling in foetal urological anomalies: when genetic counselling should be recommended, which types of genetic workup are appropriate, and which conditions indicate its use.
28.1.2.d.1. Genetic testing in the prenatal diagnosis of CAKUT
In genetic counselling, it is important to distinguish between screening and diagnostic tests. Non-invasive screening with cell-free foetal DNA (cfDNA) in maternal blood allows the detection of common aneuploidies but does not identify structural urological anomalies. When a significant ultrasound finding is present, genetic diagnosis is only possible through an invasive test that analyses foetal material directly (amniocentesis at/or beyond 15 weeks GA or chorionic villus sampling after ten weeks GA), which enables chromosomal microarray (CMA) and, when indicated, more advanced sequencing studies (WES).
Genetic evaluation forms an integral part of the management of congenital anomalies of the kidney and urinary tract (CAKUT) detected prenatally. Molecular testing refines prognostic accuracy, facilitates the identification of syndromic entities with extrarenal involvement, and provides essential information for reproductive counselling, including recurrence risk and consideration of preimplantation or prenatal testing in future pregnancies [1611].
28.1.2.d.2. Family counselling
A prenatal diagnosis of a urological anomaly imposes a considerable emotional burden on families, who frequently report anxiety and a strong need for clear, realistic prognostic information. Counselling should be structured, reiterated across several consultations, and tailored to family circumstances. Counselling must also address reproductive implications and ensure access to psychological support and written educational resources [1611-1613]. Importantly, families should be informed that prenatal findings may not always be definitive and that the working diagnosis will be verified and refined after birth. Finally, genetic counselling and multidisciplinary decision-making should always be adapted to the cultural, ethical and legal context of each country, ensuring that parental values and local regulations are fully respected in the counselling process.
28.1.2.d.3. The multidisciplinary team (MDT)
Optimal management requires a multidisciplinary team (MDT), including maternal-foetal medicine specialists, clinical geneticists, paediatric urologists and nephrologists, neonatologists and psychologists. This integrated approach ensures comprehensive evaluation and consensus-based planning. Severe LUTO represents a classical example where prognosis is uncertain and the potential for foetal intervention illustrates the complexity of decision-making, underlining the essential role of MDT discussion and referral to specialised centres [1611,1614].
28.2. Antenatal hydronephrosis
28.2.1. Definitions and follow-up during pregnancy
Antenatal hydronephrosis (ANH) is defined as a prenatally detected dilatation of the renal pelvis and is a common finding in 1-2% of pregnancies [1590,1615]. Antenatal hydronephrosis is more often seen in male foetuses compared to female foetuses at a rate of approximately 3:1 [1599,1616-1618]. The anterior-posterior renal pelvic diameter is commonly used to quantify ANH on foetal ultrasound. An APRPD that exceeds 4mm and 7mm before and after 28 weeks, respectively, is considered dilated [1619]. The ureters are considered dilated when visible [1615].
There is a large variety of clinical presentations of ANH, with a majority being transient findings (up to 80%) and others indicating more severe postnatal outcomes where surgical intervention is needed [1590]. Prenatal parameters such as parenchymal appearance, kidney size and echogenicity, and amniotic fluid volume, as well as selected foetal urinary biochemical markers (e.g. sodium, chloride, β2-microglobulin, cystatin C), may provide supportive information regarding postnatal renal function, but their predictive value remains variable [1620].
ANH has also been associated with chromosomal anomalies, single-gene disorders, and other congenital malformations, including digestive anomalies [1621]. When relevant, genetic counselling and testing should be offered based on ultrasound findings and family preferences [1622].
28.2.2. Aetiology
Antenatal hydronephrosis can present in isolation but may also be associated with abnormalities of the ureters, bladder or renal parenchyma, and is considered part of the congenital anomalies of the kidneys and urinary tract (CAKUT). Possible diagnoses that are found after birth in children with ANH include ureteropelvic junction obstruction (UPJO), duplex kidney, poly- or multicystic kidney (P/MCDK), vesicoureteral reflux (VUR), posterior urethral valves (PUV), ectopic ureter, ureterocoele and ureterovesical junction obstruction (UVJO) [1618,1623,1624]. UPJO represents the most common underlying diagnosis in cases with isolated ANH, whereas the presence of additional anomalies typically prompts more extensive postnatal investigations to clarify the underlying pathology [1625]. Figure 17 presents a proposed management pathway to differentiate between the possible diagnosis during the pre- and postnatal period [1625].
28.2.3. Classification systems for postnatal outcomes
Two classification systems are most often used to determine the severity of ANH and to support risk stratification for postnatal outcomes. The SFU grading system includes the Renal Pelvis Anteroposterior Diameter (RPAPD), collecting systems and renal parenchymal appearance [1626]. The urinary tract diameter (UDT) classification system can be used both ante- and postnatally and additionally assesses other parameters in the urinary tract, e.g. aspect of ureters and bladder compared to the SFU system [1590]. Tables 14 and 15 summarise both classification systems. A comparison between these classification systems demonstrated that both systems allow a proper risk stratification and prediction of clinical outcomes [1627].
The Hydronephrosis Severity Index (HSI), an artificial intelligence-based tool, has shown promise in predicting the presence of obstruction based on ultrasound images and SFU grading. While this may assist in decision-making regarding follow-up, its clinical use remains investigational [1628].
28.2.4. Postnatal outcomes
When ANH is diagnosed, counselling should emphasise the range of possible postnatal outcomes and the fact that prenatal findings may not be definitive. Differentiating between isolated ANH, bilateral ANH and ANH associated with other CAKUT is important for planning follow-up. Various predictive factors have been described and are now summarised. A spontaneous resolving ANH during pregnancy resulted in a recurrent postnatal hydronephrosis in 44% of children. However, none of these patients required surgical intervention during follow-up [1629].
- The Anteroposterior Renal Pelvic Diameter (APRPD) is commonly used as a marker to assess postnatal risk. In children with an isolated APRPD of < 20mm, spontaneous resolution rates of 72% have been described, with higher resolution rates for lower APRPD [1630]. Larger APRPD values and increasing measurements during gestation have been associated with a higher likelihood of postnatal intervention [1599,1618]. A cut-off of 15mm has been proposed in some studies, but no single threshold is absolute, and decisions must be guided by the overall clinical and imaging context. The presence of associated CAKUT further increases the likelihood of intervention or abnormal outcomes compared to isolated ANH [1599].
- The Urinary Tract Dilation (UTD) classification system can be used to predict postnatal outcome, and outcomes vary whether ANH is isolated or in concurrence with additional abnormalities. Resolution rates for UTD A1 of 73% and resolution rates of 21% for A2/A3 have been demonstrated [1631]. The need for additional treatment antibiotics/surgery becomes higher with increasing UDT grade [1631]. In isolated ANH, the need for surgery was shown to be lower when using the UDT classification P1 1% and P2/P3 31% [1627]. The rate of UTI was lower in the P1 group with 2% compared to 10% in P2/P3 [1627].
- The SFU classification system has been used to predict resolution and need for surgery. Grouping often occurs with grade 1-2 classified as low and grade 3-4 as high. Lower SFU grades have higher chances to resolve spontaneously and less often require surgery [1623,1632]. Spontaneous resolution rates are per grade: SFU grade 1 89-94%; grade 2 57-77%, grade 3 34.4-46% and grade 4 11% [1616,1630]. Need for surgery has been reported to be 2-15% for low grades and 32-77% for high grades, with lower ages at surgery for higher SFU grades [1616,1617,1627].
28.2.5. Long-term outcomes
Long-term follow-up studies suggest that outcomes correlate with UTD grade, but variability exists. In one nine-year follow-up study of isolated ANH, surgical intervention and UTI rates increased with UTD grade, whereas no hypertension, CKD or renal injury was observed in children with UTD P1 [810]. Children with higher grades (P2 and P3) showed higher frequencies of hypertension, CKD and renal injury, but these associations should be interpreted in the context of study heterogeneity and selection bias. A separate 13-year follow-up reported variable surgical needs and UTI rates depending on the underlying anomaly [1624].
28.2.6. Prophylactic measures against UTIs
Two systematic reviews found no proven benefits for continuous antibiotic prophylaxis in the heterogeneous group of children with ANH involving all various aetiologies. However, individual risk stratification is warranted with children with risk factors such as ureteral dilatation, circumcision status, girls and high-grade ANH [825,1633]. Circumcision was found to reduce the frequency of UTIs in boys with isolated ANH, ANH secondary to VUR and ANH secondary to PUV in a different systematic review [1634]. Additional measures, such as foreskin care and topical steroid use, may further reduce UTI risk in selected cases [1635].
Table 14: Society for Foetal Urology (SFU) classification system
| SFU grading system | |
| 0 | No hydronephrosis |
| 1 | Dilatation of renal pelvis only, with no calyceal involvement or parenchymal atrophy |
| 2 | Dilatation of both the renal pelvis and some calyces. |
| 3 | Significant dilatation of the renal pelvis and all calyces, along with a mild thinning of the renal cortex |
| 4 | Gross, balloon-like dilatation of the renal pelvis and calyces, with marked loss of the border between them and visible thinning of the renal parenchyma |
Table 15: The Urinary Tract Dilation (UTD) classification system (adopted from Nguyen et al., 2022 [1590]
| Antenatal | Postnatal (> 48h) | ||||
| UTD A1 | UTD A2-3 | UTD P1 | UTD P2 | UTD P3 | |
| APRPD | 4 - < 7mm (< 28w) 7- < 10mm (≥ 28w) | ≥ 7mm (< 28w) ≥ 10mm (≥ 28w) | 10 - < 15mm | ≥ 15mm | ≥ 10mm |
| Calyces | Any dilatation | Central dilatation | Peripheral dilatation | Any dilatation | |
| Ureter | Any dilatation (with APRPD ≥ 4mm or calyceal dilatation) | ≥ 4mm (with APRPD ≥ 10 mm or calyceal dilatation) | |||
| Parenchymal abnl., Bladder abnl., or Oligohydramnios | Yes (with APRPD ≥ 4mm or calyceal dilatation) | Yes | |||
APRPD = Anterior Posterior Renal Pelvic Diameter.
Parenchymal abnormalities = cortical thinning, hyperechogenicity, or cystic dysplasia, indistinct corticomedullary differentiation.
Bladder abnormalities = wall thickening, ureterocoele, dilatated posterior urethra.
Figure 17: Diagnostic and management pathway for foetuses with ANH (Adapted from Rickard et al., 2022) [1625] AFV = amniotic fluid volume; APRPD = anteroposterior renal pelvic diameter; CAP = Continuous antibiotic prophylaxis; HN = Hydronephrosis; HUN = Hydroureteronephrosis; SFU = Society for Fetal Urology grading system; US = Ultrasound.
AFV = amniotic fluid volume; APRPD = anteroposterior renal pelvic diameter; CAP = Continuous antibiotic prophylaxis; HN = Hydronephrosis; HUN = Hydroureteronephrosis; SFU = Society for Fetal Urology grading system; US = Ultrasound.
28.3. Congenital lower urinary tract obstruction
28.3.1. Introduction
The term congenital lower urinary tract obstruction is used to refer to intrauterine dilatation of the bladder and/or the upper urinary tract. This condition affects roughly 2-3/10,000 births [1233,1636]. The three most common causes include posterior urethral valves in males, urethral atresia or urethral stenosis [1637]. The outflow obstruction of the bladder in children with CLUTO leads to progressive bladder dilatation, bladder wall thickening, hydroureteronephrosis with subsequent compression of the renal parenchyma and/or oligo- or anhydramnios [1228]. Oligo- and especially anhydramnios can compromise pulmonary development and result in pulmonary hypoplasia, especially in or before the critical period of lung development (canalicular phase, 16-24 weeks) [1638]. The presence of CLUTO in a foetus is therefore associated with a high morbidity and pre- and perinatal mortality [1233].
Due to the heterogeneity and the rare spectrum of clinical manifestations of CLUTO, referral of such cases is recommended to a tertiary centre with multidisciplinary expertise in prenatal and postnatal management of obstructive uropathies [1228].
This section deals with the prenatal diagnosis and prenatal management of foetuses with CLUTO. Details on postnatal diagnostics, management and outcomes can be found in Chapter 21 on congenital lower urinary tract obstruction.
28.3.2. Diagnosis
28.3.2.a. Ultrasound parameters
CLUTO is usually suspected or diagnosed during the routine prenatal ultrasound check-ups. Ultrasound is the first-line diagnostic tool in foetuses with suspected CLUTO. However, in some circumstances, when technical ultrasound conditions are poor, foetal MRI may provide additional information [1247].
The following ultrasound features can help discern an obstructive pathology from isolated hydronephrosis: Megacystis, defined as a longitudinal bladder diameter of ≥ 7mm in the first trimester and ≥ 15mm in the second trimester [1228]. Bladder wall thickening, dilated posterior urethra (keyhole sign), bilateral hydroureteronephrosis [1639] and oligo- or anhydramnios [1228,1597]. However, differentiation between obstructive and nonobstructive aetiologies on prenatal US is challenging, as both have a similar sonographic appearance [1245].
Oligohydramnios is defined (beyond 16 weeks GA) as the deepest vertical pocket of amniotic fluid ≤ 2cm, anhydramnios as complete absence of amniotic fluid [1597]. Anhydramnios before 20 weeks gestation is a strong predictor of pulmonary hypoplasia and of foetal and neonatal death with survival rates of just 15-24% in this group [1228]. Therefore, the GA at the appearance of oligo- or anhydramnios is an important predictive factor [1597].
28.3.2.b. Biomarkers
As an addition to ultrasound parameters, foetal urinary biomarkers can be measured. This measurement is usually performed as a serial vesicocentesis. The idea behind this being that the first puncture most likely represents old urine, whereas the sampling done 24 and 48 hours later represents newer urine and offers an idea of bladder cycling. Common elements evaluated in foetal urine chemistries include sodium, chloride, calcium, protein, osmolality and beta-2 macroglobulin, with normal reference values shown in Table 16.
Additionally, foetal serum beta-2 microglobulin appears to have a high sensitivity (80-100%) and specificity (66-99%) in predicting postnatal renal function [1640], particularly in late gestation. As it does not cross the placenta, foetal serum beta-2 microglobulin represents foetal renal function without the influence of maternal renal function and is not dependent on gestational age [1641]. Studies have shown that beta-2-microglobulin levels above 5mg/L appear to be a predictor of postnatal renal impairment [1640-1642]. However, it cannot be used to determine the benefit of foetal intervention with regards to improvement of renal function [1637].
More recently, a 12-peptide signature (12 PUV) has been designed by proteome analysis of foetal urine. It specifically identifies foetuses who would develop ESRD before the age of two years. The final validation of the 12 PUV signature is currently ongoing [1643]. Foetal urinary peptides to predict postnatal outcome of renal disease in foetuses with posterior urethral valves (PUV) [1643,1644].
Table 16: Favourable foetal urine chemistry (reproduced from Menchaca et al., 2024) [1637]
| Foetal urinary marker | Value |
| Sodium, mEq/L | < 100 |
| Chloride, mEq/L | < 90 |
| Calcium, mg/dL | < 8 |
| Protein, mg/dL | < 20 |
| Osmolality, mOsm/L | < 200 |
| Beta-2 microglobulin, mg/dL | < 6 |
28.3.3. Management
28.3.3.a. Indication for Treatment and Staging systems
Goals of foetal intervention in CLUTO include improvement of foetal and neonatal survival, as well as improvement of postnatal kidney and bladder function. Before considering foetal intervention, life-limiting genetic or structural anomalies should be excluded [1597].
Indications for foetal intervention in CLUTO include ultrasound findings suggestive of obstruction and reduced amniotic fluid volume. Additional factors such as bladder refill and renal biochemistry can be taken into consideration for decision-making [1597].
Multiple staging systems for the estimation of the severity of the prenatal obstruction and guidance on indicating prenatal intervention have been proposed. However, there is currently no recommendation to adopt any one of these staging systems into clinical practice yet [1597].
The staging system proposed by Ruano et al. includes ultrasound parameters and foetal urinary biomarkers. The proposed system includes four stages of LUTO severity. Stage II and III foetuses are potential candidates for foetal intervention. Stage IV represents foetal renal failure leading to anhydramnios and pulmonary hypoplasia. No foetal intervention is recommended, and further studies are needed to determine the role of amnioinfusion in this population [1252].
The staging system proposed by Fontanella et al. is based solely on ultrasound parameters and uses amniotic fluid volume (AFV) and bladder volume (BV) to determine severity of foetal LUTO. LUTO cases with a BV ≥ 5.4cm3 or abnormal AF before 20 weeks’ gestation were defined as severe and those with BV < 5.4cm3 and still normal AF at the 20 weeks’ scan were defined as moderate. Risk of perinatal mortality significantly increased according to the stage of severity [1645].
Table 17: Staging systems for the estimation of the severity of CLUTO
| Ruano et al. 2017 | Stage I | Stage II | Stage III | Stage IV |
| Ultrasound | Normal AFI. No renal cysts or dysplasia | Oligohydramnios, severe bilateral hydronephrosis. Absent cysts or dysplasia | An- or oligohydramnios. Hyperechogenic kidneys, renal cysts; and/or dysplasia | Anhydramnios and anuria after monitoring bladder refilling rate. Renal dysplasia and hyperechogenicity |
| Biochemistry | Favourable after sequential sampling | Favourable after maximum of 3 sequential samplings | Unfavourable after sequential sampling | Unfavourable biochemistries and documented anuria |
| Possible foetal therapies | Weekly US monitoring | Cystoscopy or VAS | VAS with possible Amnioinfusion | Amnioinfusion |
| Fontanella et al. 2021 | Stage I | Stage II | Stage III | |
| Ultrasound | Normal AFV at 26 weeks | Bladder volume < 5.4cm3 and/or normal AFV at 20 weeks | Bladder volume ≥ 5.4cm3 and/or oligohydramnios or anhydramnios before 20 weeks | |
AFI = Amniotic fluid index; AFV = amniotic fluid volume; US = Ultrasound; VAS = Vesicoamniotic shunting..
Figure 18: Management pathway for foetuses with suspected CLUTO (as suggested by Mustafa et al.) [1597]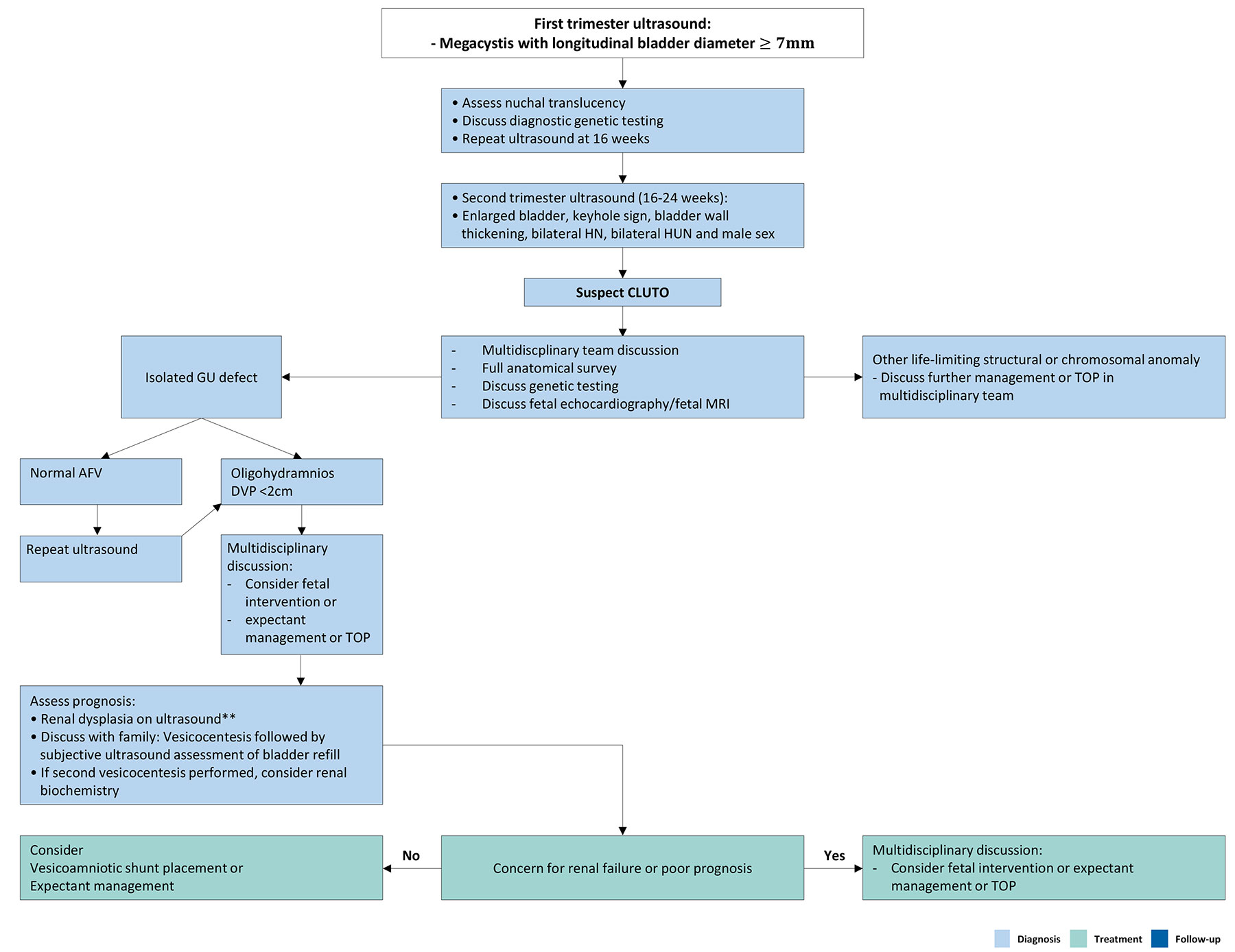 AFV = amniotic fluid volume; CLUTO = Congenital lower urinary tract obstruction; DVP = Deepest vertical pocket; GU = Genitourinary; HUN = Hydroureteronephrosis; MRI = Magnetic resonance imaging; TOP = Termination of pregnancy.
AFV = amniotic fluid volume; CLUTO = Congenital lower urinary tract obstruction; DVP = Deepest vertical pocket; GU = Genitourinary; HUN = Hydroureteronephrosis; MRI = Magnetic resonance imaging; TOP = Termination of pregnancy.
• Timing of intervention
Foetal intervention can be performed in selected cases with moderate and severe CLUTO, ideally before 27 weeks of gestation [1228]. Historically, the ideal timeframe was said to be 16-27 weeks, which represents a vulnerable phase during lung development (canalicular phase).
However, newer studies suggest that early intervention in the first trimester (< 14 weeks) is feasible, though technically more challenging and might better preserve renal function. The indication for performing an early intervention in published case series is foetal megacystis ≥ 15mm in the first trimester. Late intervention performed > 17 weeks is thought to improve survival and postnatal pulmonary function but cannot prevent kidney damage [1646].
• Techniques
The most common techniques for foetal intervention in CLUTO include vesicoamniotic shunting (VAS), foetal cystoscopy and serial amniotic infusions.
• Vesicoamniotic shunting
The most common intervention in foetuses with CLUTO is vesicoamniotic shunting (VAS). In VAS, the foetal bladder is punctured under ultrasound guidance. A drainage catheter is then introduced with one end in the bladder and the other end in the amniotic cavity [1228]. Various stenting systems are available, subject to clinician preference. The rationale of VAS is to bypass the obstruction and ensure amniotic fluid cycling by continuous drainage of the bladder into the amniotic cavity. This prevents oligo- and ahydramnios and subsequent pulmonary hypoplasia [1647].
An SR and meta-analysis on the outcomes of ‘late’ foetal interventions (16-28 weeks) including 10 studies showed an improved perinatal survival of the VAS group compared to the control group (57% vs. 38.8%) [1256,1263]. Subgroup analysis showed that perinatal survival was improved in foetuses with unfavourable urine chemistry, however, not in those with favourable urine chemistry.
The studies reporting on postnatal renal function showed a trend for a higher postnatal renal function in the VAS group compared to the conservative group at six months to two years postnatally [1256].
Newer studies suggest that early intervention before 16 weeks might achieve a higher rate of normal renal function and pulmonary function peri- and postnatally compared to later treatment [1257]. Supporting this notion are case series that show a higher overall survival rate, reported as 74% [1646], and a higher rate of foetuses with preserved perinatal renal function in 51% [1648]. However, a higher proportion of complex urethral pathologies was found in the early shunting group, posing new clinical challenges in the postnatal period [1649].
Procedure-related complications of VAS are very high with reported rates of up to 40%. Most common complications include shunt dislocation or retraction, shunt blockage, foetal ascites and premature rupture of membranes [1263]. Surgical removal of shunts was necessary in more than half of newborns after early shunt placement due to shunt migration [1650,1651].
28.3.4. Foetal cystoscopy
Foetal cystoscopy with laser ablation of the PUV has been reported as an alternative treatment to VAS. Two SR on this topic show improved survival in the treatment group comparing to the group without treatment [1256,1652]. However, numbers are small and urological fistulas have been reported in up to 10% of patients. The evidence on foetal cystoscopy is limited to case series with a limited number of patients. Therefore, no clear recommendations can be made.
28.3.5. Serial amniotic infusions
Serial amnioinfusions were first described in the 1990s in the setting of preterm premature rupture of the membranes (PPROM) [1653]. However, the role of serial amnioinfusions in foetuses with LUTO remains experimental. A recent expert consensus statement recommended using serial amnioinfusions only experimentally as part of research protocols, such as the RAFT trial [1654], as its benefits have not yet been proven.
28.3.6. Outcomes postnatally
There is a scarcity of studies on long-term outcomes after foetal intervention for CLUTO. An SR shows that postnatal renal function between six months and two years appears to be improved in patients after prenatal intervention compared to patients with no intervention [1256,1263]. After early VAS, patients appear to have higher preservation rates of renal function, even after intermediate follow-up of 4-10 years [1648].
28.3.7. Postnatal management
Details on postnatal diagnostics and management and outcomes can be found in Chapter 21 on the congenital lower urinary tract obstruction.
28.3.8. Complex urogenital pathologies - prenatal management and considerations
• Megacystis
Foetal megacystis is defined as an abnormally enlarged foetal bladder if diagnosed in the first trimester when the longitudinal bladder diameter exceeds 7mm and in the second/third trimester when the large and full bladder fails to empty during an extended ultrasound examination. The reported incidence of foetal megacystis is approximately 1 in 1,500-7,000 pregnancies, with a slight male predominance. The condition may represent a transient, self-limiting finding, or be the first manifestation of significant lower urinary tract obstruction (LUTO), chromosomal anomalies or syndromic disorders, thereby influencing prognosis, counselling and management. However, and even in the event of an isolated first-trimester foetal megacystis, completely resolved, the prevalence of chromosomal abnormalities was found in 2.4% of the cases, as was found in a large retrospective multicentre study [1655]. Nevertheless, the authors warrant for specialised paediatric follow-up. While posterior urethral valves (PUV) constitute the most common cause of LUTO in male foetuses, not all cases of foetal megacystis are attributable to PUV. Distinction is critical, as megacystis may also arise from non-obstructive aetiologies or other obstructive mechanisms (e.g. urethral atresia, prune belly syndrome). Therefore, careful differentiation between isolated megacystis and valve-related bladder outlet obstruction is essential for appropriate prenatal risk stratification and postnatal follow-up. A large bladder, bilateral hydroureteronephrosis and bladder wall thickening are the accurate predictors of LUTO [1639]. The keyhole sign, however, has a low diagnostic performance and a low specificity.
A retrospective study in 53 foetuses with a megacystis and an attempt for treatment via intrauterine vesicoamniotic shunting has concluded that, despite early intervention, no improved rate of normal renal function could be observed, and morbidity and mortality also remained high [1656].
Megacystis can be part of a syndrome known as megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS), and the large bladder is characterised as a bladder distention without mechanical obstruction [1608].
• Bladder exstrophy-epispadias complex
The bladder exstrophy-epispadias complex (BEEC) is a rare congenital anomaly involving the lower abdominal wall, urinary tract, external genitalia and, in some cases, the gastrointestinal tract. BEEC encompasses epispadias and classic bladder exstrophy. The estimated prevalence is 1:30,000-50,000 live births, with a male predominance. The aetiology is multifactorial, with contributions from genetic factors and early embryonic maldevelopment of the cloacal membrane.
Prenatal diagnosis is increasingly possible and important thanks to prenatal ultrasound and foetal MRI, enabling parental counselling and delivery planning. However, in a recent study of more than 280 children born with BEEC, only 60% had been detected during a foetal ultrasound [1606]. Another large study observing the detection during foetal ultrasound over a 20-year period revealed an increase in detection over the years [1605]. Bladder exstrophy has typical characteristics during foetal ultrasound, such as a nonvisible foetal bladder, a low insertion of the umbilical cord, normal kidney anatomy, normal amount of amniotic fluid, an abnormal pubic diastasis, a low umbilical cord insertion-to-genital tubercle length, and malformation of the external genitalia, and can be detected between the 15th and 33rd week of gestation [1607]. An early detection allows for appropriate parental counselling and time for reflection, and in some cases, termination of pregnancy. It is not always possible during foetal ultrasound to distinguish between BEEC and cloacal exstrophy, which a foetal MRI is able to do.
Despite advances in reconstructive surgery, BEEC has lifelong consequences for urinary continence, sexual function, fertility and quality of life, requiring multidisciplinary long-term follow-up.
• Prune belly syndrome
Prune belly syndrome (PBS) is another rare congenital disorder defined by the triad of deficient or absent abdominal wall musculature, bilateral cryptorchidism, and urinary tract dilation. PBS has an incidence of one in 30,000-50,000 live births and shows a male predominance in over 95% of cases [1657]. The clinical spectrum is highly variable, ranging from severe, often lethal presentations with pulmonary hypoplasia and early renal failure, to milder phenotypes compatible with long-term survival but associated with chronic urological morbidity. The prognosis of prune belly syndrome is determined by the degree of renal impairment and associated anomalies. The differential diagnosis from a bladder outlet obstruction, as in LUTO during foetal development, is challenging.
In a study with 45 prune belly syndrome patients observed over a period of 18 years, the diagnosis was made prenatally in 39%, two out of three of whom underwent an intrauterine vesicoamniotic shunt [1609]. The neonatal mortality rate was 27%, most often due to pulmonary complications. The advantage of intrauterine vesicoamniotic shunting is limited.
• Cystic kidney disease
Cystic kidney disease in the foetus refers to a heterogeneous group of disorders characterised by the presence of one or multiple renal cysts, which may be unilateral or bilateral, isolated or part of a syndromic condition. The overall incidence is estimated at approximately one in 1,000-2,000 pregnancies, with wide variability depending on the underlying diagnosis. Prenatal detection is typically achieved through routine second-trimester ultrasound, which may reveal hyperechogenic kidneys, loss of corticomedullary differentiation and variable cystic patterns. The main differential diagnoses include multicystic dysplastic kidney (MCDK), autosomal recessive polycystic kidney disease (ARPKD), autosomal dominant polycystic kidney disease (ADPKD) and syndromic ciliopathies (e.g. Meckel-Gruber syndrome). The diagnosis relies on detailed sonographic assessment, consideration of associated anomalies and, in selected cases, genetic and parental testing is advised [1658]. Genetic testing is performed in study environment [1659].
Foetal intervention is generally not indicated except in rare situations with severe oligohydramnios and preserved renal function, though evidence remains limited [1610]. Prenatal counselling should address diagnostic uncertainty, potential progression and postnatal implications, with long-term outcome depending largely on the underlying disorder, residual renal function and extra-renal involvement [1660]. In the event of a unilateral MCKD a contralateral compensatory growth of the kidney seems to be a favourable outcome parameter [1602].
28.3.9. Summary of evidence and recommendations for foetal urology
| Summary of evidence | LE |
| Ultrasound examination in the first and second trimester remain the cornerstone for detection of urinary tract malformations. | 1b |
| Megacystis is defined as a longitudinal bladder diameter of ≥ 7mm in the first trimester and ≥ 15mm in the second trimester. | 2a |
| An APD of ≥ 4mm before 28 weeks and ≥ 7mm after 28 weeks is considered abnormal. An APD > 15mm strongly predicts the need for postnatal surgery. | 1b |
| Individual risk stratification regarding the need for antibiotic prophylaxis is warranted in children with risk factors such as ureteral dilatation, circumcision status, being a girl and showing high-grade ANH | 4 |
| Early oligo- and especially anhydramnios can compromise pulmonary development and result in pulmonary hypoplasia. | 1b |
| Ultrasound parameters that help differentiate obstructive from nonobstructive pathologies include: megacystis, bladder wall thickening, dilated posterior urethra (keyhole sign), bilateral hydroureteronephrosis and oligo- or anhydramnios. | 4 |
| Anhydramnios before 20 weeks’ gestation is a strong predictor of pulmonary hypoplasia and of foetal and neonatal death. | 2a |
| VAS for CLUTO has been shown to improve perinatal survival in select patients. | 4 |
| Differentiation between isolated megacystis and valve-related bladder outlet obstruction is essential for appropriate prenatal risk stratification and postnatal follow-up. | 4 |
| Recommendations | Strength rating |
| Perform screening ultrasound examination in the first (11-14 weeks) and second (17-22 weeks) trimesters to assess for urinary tract malformations. | Strong |
| Care for patients with urinary tract malformations in a multidisciplinary team (MDT), including maternal-foetal medicine specialists, clinical geneticists, paediatric urologists and nephrologists, neonatologists, and psychologists. | Strong |
| Obtain foetal urine for prenatal biochemistries by means of serial vesicocenteses. | Weak |
| Exclude life-limiting genetic or structural anomalies before considering foetal intervention in congenital lower urinary tract obstruction. | Weak |
| Consider foetal intervention in a foetus with ultrasonographic signs of obstruction and reduced amniotic fluid in the second trimester. | Weak |